






Abstract
Patients with Turner syndrome are prompt to develop spontaneous acute aortic dissection following insidious aortic dilatation, with abnormal cardiovascular anatomy and consequently require specific guidelines for regular surveillance since they represent a subset of high-risk young patients. We report a rare and uncommon case of spontaneous acute aortic dissection in a 48-year old female patient with Turner syndrome who was not apparently eligible for a prophylactic surgery. A CT scan showed a Stanford type A aortic dissection and was urgently referred for surgical management. We operated on the patient under deep hypothermia (18°C) and circulatory arrest with a retrograde cerebroplegia as the primary entry tear was located in the arch. The postoperative course was uneventful and the patient was discharged at the eighth postoperative day. Following description of this case, special attention was paid to determine predisposing risk factors for aortic dissection to be specifically adjusted to TS patients.
INTRODUCTION
Patients with underlying connective tissue diseases are predisposed to life-threatening aortic complications such as aneurysms, dissections and spontaneous ruptures. The treatment of genetically triggered aortic lesions is demanding and may be associated with significant rates of morbidity, mortality and iterative surgical procedures.
Classification of disorders of the connective tissue is complex and includes those well known to be caused by fibrillin mutations (Marfan syndrome), collagen mutations (Ehlers–Danlos syndrome) and transforming growth factor-β receptor mutations (Loeys–Dietz syndrome), but also those in which a connective tissue disorder is hypothesized but not proved, such as bicuspid aortic valve syndrome and Turner syndrome.
We report a case of spontaneous acute aortic dissection in a Turner syndrome (TS) patient who was not apparently considered at risk for aortic dissection or rupture.
In addition, we propose a comprehensive review of the literature to validate specific criteria adjusted to TS patients for monitoring aortic dimensions and indications for a prophylactic surgery.
MATERIALS AND METHODS
Case
A 48-year old woman with TS presented with a spontaneous acute aortic dissection.
The height and the weight of the patient were, respectively, 155 cm and 67 kg with a body surface of 1.66 m2.
She was regularly followed up for a ‘moderate’ and stable aortic dilatation (diameter 40–42 mm) with no aortic valve regurgitation. Therefore, she was not identified for a prophylactic intervention.
In addition, she had a medical history of moderate hypertension treated by beta-blockers.
After waking up, the patient called the emergency department for a moderate but persistent chest pain that started 3 days earlier. After admission in hospital, she underwent a CT scan that disclosed a Stanford type A aortic dissection without any pericardial effusion. The primary entry tear was located in the arch (Fig. 1).
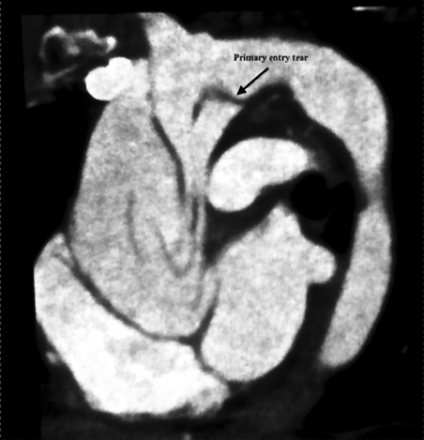
CT scan shows aortic dissection with a primary entry tear located in the concavity of the arch.
Cardiac echography showed a moderate central aortic insufficiency (grade 2) with a tricuspid aortic valve.
She was then referred to our institution for surgical management.
Surgical technique
Because of the primary entry tear located in the arch, and in order to explore and repair the arch according to an ‘open-suture’ technique, deep hypothermia with circulatory arrest was decided.
The strategy to protect the brain was a crucial step because of potentiality of both arterial and venous major anomalies as described in Turner syndrome. Then, after discarding aortic coarctation, persistent left superior vena cava (LSVC) and partial anomalous pulmonary venous return (PAPVR) to the left brachiocephalic vein, we opted for a retrograde cerebroplegia under deep hypothermia (18°C) and circulatory arrest.
After cannulation of the femoral artery, surgical access was gained through a full sternotomy. No haemorragic pericardial effusion was noted. Ascending aorta was dissected and measured at 44 mm. Hypothermic CPB was established after cannulation of the right atrium. Myocardial protection was achieved by retrograde cardioplegia.
After cross-clamping, the ascending aorta was resected immediately above the coronary ostia and the aortic root was repaired with GRF glue. Measurements of the annulus, Valsalva sinuses and sinotubular junction were, respectively, 21, 24 and 23 mm.
Aortic valve was carefully inspected and each cusp was resuspended. The non-coronary leaflet was moderately elongated and was corrected by a subcommissural plication to reduce the length of its free margin. Finally, a valve-sparing root replacement was not required and a 28-mm Intergard® prosthesis was proximally sutured and aortic valve competence was checked.
After reaching 18°C, retrograde cerebroplegia and circulatory arrest were set up and the aortic clamp removed. Careful inspection of the arch showed a primary entry tear located in the proximal segment of the concavity of the arch.
The posterior wall of the arch was extremely thin and friable and required reinforcement with a complete inner and outer felt Teflon fixed with separate U-stitches.
Then, distal anastomosis of the prosthesis was completed with running sutures of 5–0 Prolene. The arch and the upper portion of the prosthesis were de-aired by slowly restarting the arterial femoral line. The prosthesis was clamped and the retrograde perfusion stopped. A cannula was inserted in the prosthesis above the clamp and CPB was antegradely restarted to re-warm the patient. Before removing aortic cross-clamp, and after de-airing the heart, a warm myocardial reperfusion was achieved.
Weaning from CPB was uneventful. Cross-clamping and circulatory arrest times were, respectively, 112 and 57 min. Recovery was uneventful and the patient was discharged on the eighth postoperative day.
Postoperative echocardiography showed a perfect competent valve without any residual leakage.
Histology showed cystic medial necrosis (Fig. 2a) with intense mucoid degeneration (Fig. 2b), without any fragmentation or separation of elastic fibres.
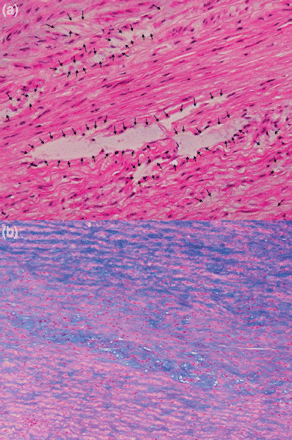
(a) Cystic medial necrosis (black arrows) without any fragmentation or separation of elastic fibres. (haematoxylin and eosin staining; original magnification ×200). (b) Intense mucoid degeneration (blue areas) located in the medial layer (Alcian blue staining; original magnification ×100).
DISCUSSION
Turner syndrome (TS) is a chromosomal disorder affecting ± 1/2500 live-born females [1, 2]. Initially described in 1938 by Henri Turner [3], it is characterized by a complete or partial loss of the second sex chromosome resulting in a complete or partial monosomy for the X chromosome with or without cell line mosaicism.
Multiple clinical features have been described and usually associate short stature with gonadal dysgenesis, renal malformation (solitary kidney), facial anomalies and pterygium colli [4].
However, the most serious clinical presentation of the syndrome is due to congenital cardiovascular lesions whose incidence varied from 17% [5, 6] to as high as 45% [7]. Price and Wilson [8] clearly demonstrated that cardiovascular abnormalities in TS are directly responsible for a higher mortality rate (×3) with a significantly reduced life expectancy (13 years) compared with the standard population. Additionally, risk of developing specific cancers such as gonadoblastoma, meningioma, brain tumours and neoplasm of uterus is significantly increased.
Independently of anatomic cardiovascular anomalies, TS patients have smaller and hypertrophic left ventricular cavities, with an increased left ventricular mass, mainly due to hypertension that occurs in 50% of TS [9].
The cardiovascular phenotype in TS mainly captured valve abnormalities (20%) like bicuspidy (17.5%) [7], aortic coarctation (5–12.5%) and other forms of left-sided obstructions.
In a cohort of 85 adults with TS, Ho et al. [10] reported elongation of the transverse arch (49%), aortic coarctation (12%) and abberant right subclavian artery (8%).
Finally 1–2% have septal defects or mitral valve prolpase [6].
Additionally, Ho et al. [10] disclosed major thoracic venous anomalies including persistent LSVC (13%) and PAPVR (13%). The most common feature of PAPVR arose from the upper lobe to the left brachiocephalic vein.
LSVC was significantly associated with elongation of the transverse arch and with an aberrant right subclavian artery.
Ho et al. [10] showed that neck webbing and increased thoracic anterior-to-posterior dimension diameters were strong predictors for arterial and venous anomalies.
Because of their specific short stature, usual thresholds to define aortic dilatation in Marfan syndrome cannot be applied to TS.
In Marfan syndrome, guidelines for monitoring of aortic root dimension and indications for surgery to prevent dissection and treat aortic valve disease are well known.
Aortic dissection was traditionally considered as a rare complication in TS with only 85 cases reported between 1961 and 2006 [11].
However, recent publications [12, 13] reported in TS a high rate of aortic dissections, including patients without predisposing factors such as bicuspid aortic valve emphasizing the direct role of the vascular wall to set up conditions for spontaneous acute aortic dissection.
Therefore, incidence both of aortic dilatation and dissection has probably been underestimated.
Multiple factors may explain this bias of analysis:
Marfan syndrome were prominent and focused interest to validate guidelines for treatment, whereas treatment guidelines for other disorders of connective tissues were lacking.
Knowledge of the natural history and clinical findings of Marfan syndrome have been used as guidelines regarding proper monitoring and treatment of TS individuals with aortic dilatation.
In TS, it remains unclear whether aortic dissection is preceded by progressive dilatation of the aortic root as it is in Marfan syndrome.
The literature on aortic dissection of women with TS was sparse and often consisted in sporadic cases reporting acute aortic dissection.
Until recently, there was no published comprehensive review of aortic dissection in TS, making it difficult to validate accurate guidelines regarding monitoring and treatment.
Consequently, in TS, the medical community has been slow to validate evidence for a significant risk of acute aortic dissection and dismal prognosis.
Thresholds of 50 mm for the general population and 45 mm for those with Marfan syndrome are usually used for guiding prophylactic surgery to treat the aneurysmal segment [14]. However, it has been strongly suggested that surgery at a 45-mm diameter would save more lives [15, 16].
Nevertheless, no specific data were recorded to know whether these thresholds could be safely considered for TS.
Because of their small stature (ascending aortic diameters <50 mm) patients with TS are rarely considered candidates for a prophylactic intervention.
Matura et al. [17] studied a cohort of 166 TS patients (average age: 36.2 years) and clearly showed that the 50-mm threshold is used and adapted for a normal-sized adult. On the other hand, the 50-mm threshold is likely too high for TS patients as they are typically <147 cm in height.
Then, in order to improve inclusion of TS patients for prophylactic aortic surgery and risk for aortic dissection, Matura et al. [17] proposed to adjust the aortic diameter to the body surface area and defined an aortic size index (ASI).
In the cohort of 166 patients, 25% of TS women with absolute ascending aortic diameter >35 mm and 33% of those with ASI >25 mm/m2 experienced aortic dissection over the 3-year of follow-up.
Matura et al. [17] proposed that individuals with a dilated ascending aorta defined as ASI >20 mm/m2 require close cardiovascular surveillance, while TS patients with an ASI >25 mm/m2 are significantly at the highest risk for aortic dissection.
In TS patients, ASI is a reliable criterion to the predicted risk for aortic dissection, and therefore to consider prophylactic surgery.
Usually aortic dimensions before the dissection or rupture are rarely available. In our case, the patient underwent regular close cardiologic surveillance and retrospectively we could obtain data about aortic dimensions.
During regular follow-up, diameters of the ascending aorta were found stable at 42 mm (transversal diameter) and 44 mm (anterior-to-posterior diameter). Therefore, resulting from usual criteria used for both Marfan syndrome and patients with bicuspid aortic valve, our patient was not apparently eligible for a prophylactic surgery.
A similar apparent ineligibility was reported by Jagannath et al. [18] with a TS patient successfully operated for an acute aortic dissection and in whom the aortic diameter was 35 mm.
Nevertheless, if we had considered the ASI as a predictor of risk for acute aortic dissection, the patient would have been eligible for a prophylactic surgery, with the ASI calculated at 25.3 mm/m2 (transversal) and 25.1 (anterior-to-posterior).
Histological evidence of cystic medial necrosis in the aortic wall sampled from our patient is similar to that found in Marfan syndrome or in patients with bicuspid aortic valve and suggests a common tissular expression despite a genetically diverse background [19, 20].
Nevertheless, in Marfan syndrome, aortic histology shows separation and fragmentation of elastic fibres associated with cystic medial necrosis, whereas, in our patient, the aortic histology showed cystic medial necrosis, however, with no evidence of either fragmentation separation or atrophy of elastic fibres.
In TS, an intense mucoid degeneration is observed in the medial layer.
In addition, our patient had neither atheromatous histological evidence nor wall thickening, resulting in apparent and extreme friability of the aortic wall mainly located in the posterior concave segment of the arch, where the primary entry tear was found.
Therefore, we had to achieve a specific and cautious reinforcement of the posterior wall of the arch.
Unlike Ostberg et al. [21], who recently documented intimal-medial thickening and vessel dilatation diffusely in the vasculature, we did not observe any similar anomalies.
Finally, overall knowledge of Turner syndrome remains unclear and incomplete, and this review reinforced the fact that, still, very little is known about this physiopathological entity and mechanisms of both aortic dilatation and dissection.
To optimize surveillance of TS patients and improve life expectancy, the Turner syndrome NIH Consensus Study Group published focused guidelines [22] and promoted education of both physicians and patients [23].
This education is highly necessary because clinical presentation of acute aortic dissection in TS is often uncommon with an intermittent and prolonged chest pain that may last several days before rupture. The pain is frequently reported as moderate. Our patient had such a clinical insidious presentation as also similarly reported by Carlson and Silberbach [11].
Our patient clearly demonstrated a serious failure in the predictive analysis of risks for lethal complications, only because both the medical community and the patient had an obvious lack of knowledge and information.
To gain a better understanding of aortic dissection in females with TS and anticipate risks of severe complications, an International Turner Syndrome Aortic Dissection Registry (ITSADR, http://www.tissus.org/readweb.asp?wid=3092) has been created.
This database collects information to profile specific risk factors for aortic dissection and gain a better understanding of the natural history of aortic dilatation that leads to spontaneous acute dissection or rupture.
In conclusion, anatomic criteria resulting from Marfan syndrome are not reliable for TS, since spontaneous aortic dissection occurs even when the ascending aorta is less than 45 mm and even, in several cases, when the aorta is less than 40 mm.
Therefore, specific thresholds for a prophylactic aortic surgery must be specifically adjusted to TS and probably to patients with a short stature and free from of connective tissue disorders.
Regarding Turner syndrome, there is a fundamental interest to index the aortic diameter to the body surface (ASI), as nearly all individuals have a short stature and common manifestations of the TS phenotype.
ASI is particularly important for physicians who follow up women with TS or patients with short stature, since TS patients have to be aware of a high risk for spontaneous aortic dissection that is significantly increased particularly during pregnancy and while undergoing assisted reproductive techniques. This latter point is crucial as many Turner patients are candidates for such techniques.
If criteria to include TS patients for prophylactic aortic surgery are not clearly established, then TS patients with ASI >25 mm/m2 or diameter >35 mm are at highest risk for aortic dissection and should be referred for prophylactic surgery to avoid life-threatening complications.
Specific education of both TS women and physicians is mandatory, and should focus on risks during pregnancy and signs that may herald both an aortic dissection and dilatation, as in many of the reported cases, the symptoms developed insidiously over the course of hours to days.
Conflict of interest: none declared.
REFERENCES
Author notes
In memory of Pr Jean-Pierre Carteaux.

{kind=link}
{kind=link}